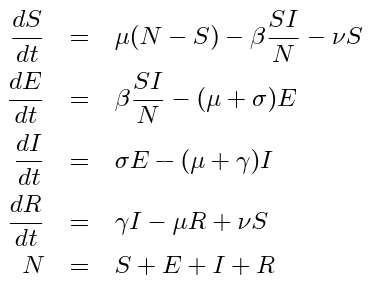W naszym ostatnim i zarazem największym zadaniu zaliczeniowym odejdziemy od tematyki epidemiologicznej i zajmiemy się przetwarzaniem obrazów i ich kompresją. Konkretnie, naszym zadaniem będzie napisanie programu pozwalającego na “ukrywanie” informacji w obrazach, czyli steganografię. Wykorzystamy najprostszą metodę steganografii, czyli wykorzystanie modyfikacji najmniej znaczącego bitu do zakodowania “ukrytej informacji”.
Zasada działania:
Załóżmy, że mamy do przekazania komuś wektor bitów S, długości N (może to być obraz, lub tekst – jest to dla nas w tej chwili bez znaczenia). Możemy wziąć jakiś obraz reprezentowany w standardowym formacie RGB, z 8 bitami na kanał i powiedzmy wymiarach 2000×1000 pikseli. Obraz ten, możemy wczytać do pamięci w postaci macierzy M o wymiarach 2000x1000x3 z typem danych int8uint8. Zauważmy, że każda z liczb w tej macierzy zawiera intensywność jednego z kolorów podstawowych w postaci wartości od 0 do 255. Niewielkie zmiany tych wartości (np. o 1) nie są łatwo postrzegalne dla ludzkich oczu, ale program komputerowy może bardzo łatwo je wykryć.
Nasze zadanie będzie polegało na tym, że każdy kolejny bit z wektora S “wstawiać” jako ostatni bit kolejnego bajtu macierzy M. Jak go wstawiać? otóż wystarczy, że sprawdzimy czy reszta z dzielenia kolejnego bajtu macierzy M przez 2 jest równa wartości odpowiedniego bitu z wektora S.
M.reshape((2000*1000*3,))[i]%2 == S[i]
Jeśli tak, to nic nie musimy robić. Jeśli nie, to musimy odjąć lub dodać 1 do M.reshape((2000*1000*3,))[i], żeby nasze równanie było prawdziwe (uważając przy tym aby nie odjąć od zera ani nie dodawać do 255).
W ten sposób otrzymujemy nową macierz M, która ma tę własność, że ostatnie bity reprezentacji kolejnych bajtów M są równe kolejnym bitom S. Taki plik możemy zapisać jako obraz i będzie on bardzo subtelnie wizualnie zmieniony, natomiast odbiorca tego pliku, który wie czego szukać może bardzo łatwo odczytać wektor S
Aby rozwiązanie było praktyczne, musimy jeszcze być w stanie podać długość wektora S oraz typ danych do zapisu tego wektora. W naszym rozwiązaniu, pierwszy zakodowany bit (M[0]%2) będzie określał, czy mamy do czynienia z tekstem w alfabecie DNA (M[0]%2==0) czy z obrazem w skali szarości (M[0]%2==1). Kolejne 32 bity będą podawać nam długość ciągu DNA (w postaci jednej 32 bitowej całkowitej liczby dodatniej w kodzie dwójkowym) lub wymiary obrazka (w postaci dwóch 16bitowych całkowitych liczb dodatnich oznaczających odpowiednio szerokość x i wysokość y zakodowanego obrazka).
Zadanie:
Napisz moduł pythonowy (z komentarzami!) o nazwie stegano.py, który realizuje następujące funkcje:
- read_image_greyscale (image_file) – zwracający wymiary obrazka oraz ciąg bitów wartości na podstawie pliku w formacie png (2 pkt)
- write_image_greyscale (x,y,bit_vector,output_image_file) – zapisujący obrazek do formatu png w skali szarości (2pkt)
- read_DNA(text_file) – zwracający ciąg bitów na podstawie pliku tekstowego zawierającego wyłącznie alfabet ACGT (kodowanie bitowe: A=00 ,C=01, G=10, T=11) (2 pkt)
- write_DNA(bit_vector,output_text_file) – zapisuje ciąg liter DNA do pliku tekstowego na podstawie ciągu bitów wg powyższego kodowania (2 pkt)
- encode_DNA(bit_vector,image_file, output_file) – wczytujący obraz RGB z pliku image file i zakodowujący wektor bit_vector reprezentujący DNA w ostatnich bitach obrazu (zgodnie z powyższym opisem, łącznie z 33 bitami nagłówka). Zmodyfikowany obraz powinien być zapisany do pliku output_file. Jeśli obraz jest za mały aby zakodować cały wektor, program powinien zwrócić błąd. (5 pkt)
- encode_image(bit_vector,x,y,image_file,output_file) – wczytujący obraz RGB z pliku image file i zakodowujący wektor bit_vector reprezentujący obraz w ostatnich bitach obrazu (zgodnie z powyższym opisem, łącznie z 33 bitami nagłówka). Zmodyfikowany obraz powinien być zapisany do pliku output_file. Jeśli obraz jest za mały aby zakodować cały wektor, program powinien zwrócić błąd. (5 pkt)
- decode(image_file, output_file) – powinien wczytywać obraz RGB z pliku, odczytywać z nagłówka (33 ostatnie bity z pierwszych 33 bajtów) informację o tym, czy zakodowane jest DNA, czy obraz, następnie odkodowywać informację i zapisać (korzystając z write_DNA lub write_image_grayscale) wynik do pliku output_file. (5 pkt)
- main(), która powinna umożliwiać zakodowywanie i odkodowywanie informacji przy pomocy parametrów z linii komend obsługiwanych przy pomocy modułu Argparse. (2 pkt)
Terminy:
Rozwiązania (w postaci e-maila z normalnym załącznikami w postaci pliku stegano.py oraz zakodowanych przykładowych danych i dopiskiem [ONA-3] w temacie) w ciągu 3 tygodni od dzisiaj, czyli do 18. VI 2020
Przykładowe dane:
Można popróbować np. zakodować taki obrazek:
lub kod DNA koronawirusa covid-19.fasta
W jakimś ulubionym pliku ze zdjęciem przedstawiającym swoją fizjonomię.
Wersja bonusowa (+5 pkt)
Można wyposażyć nasz program w dodatkową funkcjonalność polegającą na kompresji ciągu bitów przed zakodowaniem. Jeśli ktoś zaimplementuje kompresję (i dekompresję) metodą LZ77 to może otrzymać dodatkowe 5 punktów).

 Write a code that uses simplified BWT matching, based on today’s assignments, to create a BWT of the E. coli genome (
Write a code that uses simplified BWT matching, based on today’s assignments, to create a BWT of the E. coli genome (