
Naszym zadaniem będzie implementacja funkcji, która będzie wykorzystywac metodę liniowych najmniejszych kwadratów, aby znaleźć podstawową liczbę odtwarzania dla przypadków COVID-19 w Polsce.
W naszym (niezmiernie uproszczonym i nie nadającym się do zastosowań praktycznych, ale ilustrującym kluczowe idee) modelu, przyjmiemy, że początkowy rozwój wirusa w populacji rośnie wg wykładniczego wzrostu:
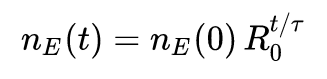
gdzie t oznacza czas (w dniach), n_E(t) oznacza liczbę osób chorych w momencie t, R_0 jest szukaną podstawową liczbą odtwarzania, zaś \tau przyjmiemy za równą 1.
Nasz program będzie wykorzystywał fakt, że to równanie po obustronnym zlogarytmowaniu staje się liniowe:
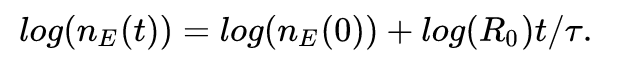
Jeśli mamy dane o liczbie potwierdzonych chorych w pewnym okresie (dla każdego kolejnego dnia), znamy \tau, to możemy wykorzystać liniową ważoną metodę najmniejszych kwadratów do dopasowania rozwiązania, aby znaleźć log(R_0).
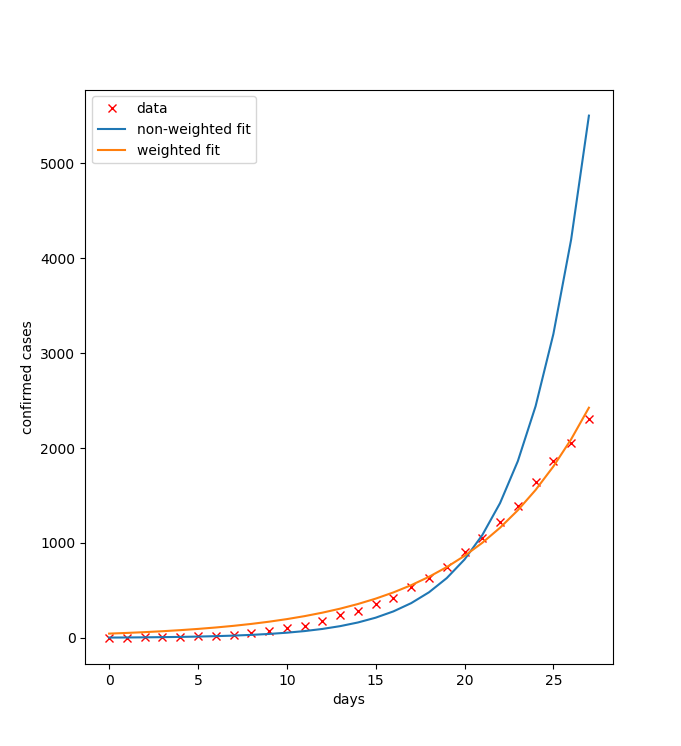
Warto pamiętać, że w tym przypadku, jeśli nie zastosujemy wag w metodzie najmniejszych kwadratów, nasza metoda będzie minimalizować |log(x)-log(b)|^2, co nie będzie prawidłowe. Jak widać na poniższym rysunku, dopasowanie do danych jest lepsze (zwłaszcza z prawej strony) dla problemu ważonego.
 Dlatego warto przypomnieć sobie zadanie o ważonym zagadnieniu liniowych największych kwadratów, które ma podane rozwiązanie w materiałach do metod numerycznych. W naszym przypadku wagi w_i są równe n_E(i).
Dlatego warto przypomnieć sobie zadanie o ważonym zagadnieniu liniowych największych kwadratów, które ma podane rozwiązanie w materiałach do metod numerycznych. W naszym przypadku wagi w_i są równe n_E(i).
Punktacja:
- Napisanie funkcji wykorzystującej funkcję scipy.linalg.lstsq (proszę nie używać funkcji typu polyfit) i obliczającej ważony liniowy problem najmniejszych kwadratów (5 pktów)
- Napisanie funkcji wykorzystującej funkcję urlopen() i dane dostępne publicznie aby pobrać pierwsze 28 dni, kiedy w wybranym kraju (np. Polsce) występowały zachorowania (tj. pomijając pierwsze zera w linii odpowiadającej danemu krajowi) (5 pktów)
- Napisanie programu, który wykorzystuje te dane, aby obliczyć R_0 zgodnie z wzorami podanymi powyżej. Program powinien też generować wykres podobny do powyższego. (5 pktów)
Rozwiązania należy nadsyłać w ciągu dwóch tygodni (czyli do 13. maja włącznie) na mój adres e-mail z dopiskiem [ONA-2] w tytule. Jak zwykle, proszę o skomentowane pliki .py jako normalne załączniki.